





27 août 2020
Nature Communications 11, Numéro d’article : 4282 (2020)
Traduction :Antilla.
La principale protéase, la Mpro (ou 3CLpro) dans le SARS-CoV-2 est une cible médicamenteuse viable en raison de son rôle essentiel dans le clivage du polypeptide du virus. La péritonite infectieuse féline, une infection mortelle à coronavirus chez les chats, a été traitée avec succès auparavant avec un promédicament, le GC376, un inhibiteur de protéase à base de dipeptide.
Nous montrons ici que le promédicament et son parent GC373 sont des inhibiteurs efficaces du Mpro du CoV-SAR et du CoV-SAR-2, avec des valeurs de CI50 de l’ordre du nanomolaire. Les structures cristallines de la Mpro du CoV-2-SARS avec ces inhibiteurs présentent une modification covalente de la Cys145 nucléophile. L’analyse par RMN révèle que l’inhibition se produit par la formation réversible d’un hémiacétal. Les GC373 et GC376 sont de puissants inhibiteurs de la réplication du SARS-CoV-2 en culture cellulaire. Ce sont de puissants candidats médicaments pour le traitement des infections à coronavirus humain, car ils ont déjà donné de bons résultats chez les animaux. Les travaux menés ici définissent le cadre de leur utilisation dans les essais sur l’homme pour le traitement du COVID-19.
Introduction
L’épidémie de COVID-19 s’est transformée en pandémie en raison de la nature virulente du SRAS-CoV-2, atteignant plus de trois millions de cas dans le monde à la fin avril 2020, le nombre de personnes infectées augmentant rapidement dans le monde entier1. Ce scénario actuel contraste avec l’épidémie de SRAS plus virulente mais moins efficacement transmise en 2002-2003, qui n’a enregistré que 8 000 cas et 774 décès2. Il existe un besoin urgent de thérapies antivirales pour les infections aiguës à COVID-19, surtout en attendant qu’un vaccin efficace soit mis au point.
Les coronavirus sont des virus à ARN qui détournent la machinerie translationnelle de l’hôte pour générer des protéines virales. L’ARN viral code pour deux polyprotéines qui se chevauchent : pp1a et pp1ab, qui sont respectivement de 450 kD et 750 kD. Les polyprotéines doivent être clivées afin de libérer des protéines fonctionnelles individuelles pour la réplication et la transcription virales. Les protéases codées virales comprennent la protéase principale (Mpro), également appelée 3CLpro, et une protéase de type papaïne (PLpro). La Mpro coupe les polyprotéines en 11 positions, principalement au niveau des séquences Leu Gln | Ser Ala Gly conservées, ce qui permet l’assemblage du complexe de réplication virale3.
Compte tenu de son rôle crucial dans la réplication du virus, la Mpro du SRAS-CoV-2 est une cible médicamenteuse de premier plan pour la thérapie antivirale COVID-19. Le coronavirus Mpro est une cystéine-protéase pour laquelle il existe de nombreuses classes d’inhibiteurs4. Les inhibiteurs de protéase sont des candidats médicaments courants s’ils répondent aux exigences de faible toxicité, de solubilité et de réversibilité5. Plusieurs protéases ont été identifiées comme cibles moléculaires et utilisées pour le développement de nouvelles classes de médicaments5, dont le Tipranavir pour le traitement du VIH6. Toutefois, l’inhibition des protéases de la cystéine par des espèces réactives au thiol est souvent intenable pour les médicaments à usage humain, à moins que l’inhibiteur ne soit réversible. Les médicaments accepteurs de Michael qui sont irréversibles in vivo, tels que le Rupintrivir, ont échoué dans les essais cliniques en raison de leur faible biodisponibilité3. Une réaction irréversible indésirable se produit avec de nombreux thiols de mammifères pour détruire l’inhibiteur. La réaction avec les thiols des protéines de l’hôte pourrait aussi potentiellement conduire à une toxicité aiguë ou à une réaction immunitaire. À cet égard, la réaction réversible des thiols avec les inhibiteurs aldéhydiques pour former des hémithioacétals présente une opportunité unique d’inhibition efficace de la cystéine protéase, car ils peuvent potentiellement se lier plus efficacement dans le site actif de leur protéine cible qu’avec d’autres thiols7. En outre, les produits d’addition hydrosolubles du bisulfite d’aldéhyde sont facilement rendus réversibles à partir de l’aldéhyde parent, et réversibles dans des conditions physiologiques8. Ces composés peuvent être des promédicaments idéaux pour l’inhibition de la cystéine protéase, comme décrit ci-dessous.
Dans les premières études, nous avons mis au point des inhibiteurs à base de peptides, notamment des aldéhydes, contre les cystéine protéases virales7 qui ont ensuite été étudiés avec Mpro lors de l’épidémie de coronavirus du SRAS (SARS-CoV) en 20039. Les aldéhydes peptidiques et leurs dérivés bisulfites ont ensuite été utilisés pour inhiber la principale protéase du coronavirus félin FCoV10. Le FCoV provoque généralement des symptômes légers, mais il peut entraîner une péritonite infectieuse féline (PIF), qui est généralement mortelle chez les chats. L’adduit du bisulfite GC376, un promédicament qui se convertit facilement en aldéhyde peptidique GC373, a été bien toléré et capable de bloquer l’infection chez les chats11. Cette étude, ainsi que d’autres études, notamment sur le coronavirus Mpro du furet et du vison, ont démontré la grande spécificité de cet inhibiteur de protéase10,12,13. Une structure cristalline de GC376 a été résolue avec le Mpro homologue du syndrome respiratoire du Moyen-Orient (MERS) et a démontré une interaction covalente avec la cystéine catalytique du Mpro14. Récemment, les structures de la protéase Mpro du SRAS-CoV-2 ont été résolues avec un inhibiteur de cétoamide à base de peptide15 et divers médicaments réadaptés tels que des agents anticancéreux16. Toutefois, ces inhibiteurs de la Mpro n’ont pas été testés sur des modèles animaux d’infection à coronavirus et n’ont pas été signalés dans des essais sur l’homme ou l’animal pour le SRAS. Un certain nombre d’inhibiteurs Mpro sont connus pour avoir des effets secondaires graves et une toxicité sur les cellules humaines, en particulier ceux qui sont des agents anticancéreux. Dans cette étude, nous examinons si les GC373 et GC376, établis comme médicaments efficaces chez les chats, inhibent le CoV-2 Mpro du SRAS de manière réversible et ont un potentiel d’utilisation comme thérapie antivirale chez l’homme.
Dans la présente étude, nous examinons l’utilisation du promédicament GC376 et du médicament GC373 pour tester l’inhibition du CoV-2 Mpro du SRAS in vitro. Nous démontrons à l’aide du CoV-2 Mpro-SARS recombinant que le GC373 et le GC376 sont de puissants inhibiteurs de l’ordre du nanomolaire. Les structures cristallines et l’analyse RMN du SARS-CoV-2 Mpro avec GC373 et GC376 démontrent que le médicament est attaché de manière covalente à Cys145 en tant qu’hémiacétal et révèle des résidus importants dans la spécificité de l’inhibiteur. En culture cellulaire, nous montrons que le GC376 et le GC373 bloquent la réplication du virus sans aucune toxicité. Cette étude montre l’efficacité du médicament félin pour une utilisation contre le SRAS-CoV-2 et un traitement possible pour le COVID-19.
Résultats
Les GC373 et GC376 inhibent le SARS-CoV-2 Mpro
Nous avons synthétisé les principaux composés dipeptidyles, l’aldéhyde GC373 et l’adduit de bisulfite GC376 (Fig. 1a)10 (Figures supplémentaires 1-12), pour tester si ces inhibiteurs du FCoV et du FIPV sont efficaces contre le Mpro du CoV-SARS-2 et le Mpro du CoV-SARS (associé à l’épidémie de 2002) qui ont des séquences d’acides aminés très similaires : 95% d’identité/98% de similarité. Ce composé se compose d’un substitut de glutamine en position P1 du substrat, d’un Leu en position P2 et d’un groupe benzyle en position P3, ce qui reflète la spécificité connue pour le Mpro du CoV-2 du SRAS. Le SARS-CoV-2 Mpro a été cloné en tant que protéine de fusion marquée SUMO, ce qui a permis une expression à haut rendement, une stabilité accrue et la génération de N- et C-terminaux natifs (Figures supplémentaires 13 et 14). De même, le SARS-CoV Mpro a été exprimé et purifié pour obtenir des N- et C-terminaux natifs selon les méthodes précédentes9. Les paramètres cinétiques pour le SARS-CoV Mpro et le SARS-CoV-2 Mpro ont été déterminés à l’aide d’un peptide synthétique FRET-substrat avec une paire donneur-accepteur anthranilate-nitrotyrosine (Abz-SVTLQSG-TyrNO2R – Tableau 1 et Fig. 15 supplémentaires) car il présente une sensibilité 10 fois supérieure à celle du système EDANS-Dabcyl équivalent17. Le SARS-CoV-2 Mpro et le SARS-CoV Mpro ont tous deux montré une liaison de substrat coopérative du substrat FRET (figure supplémentaire 16).
Fig. 1 : Les Mpro de SARS-CoV-2 et SARS-CoV sont inhibés in vitro par la GC373 et la GC376.
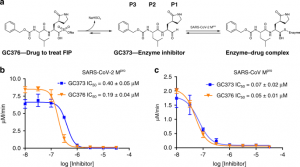
figure1
a Représentation schématique du promédicament inhibiteur GC376, utilisé pour traiter les chats atteints de PIF, et GC373, l’inhibiteur de protéase actuel. b Valeurs de la CI50 pour GC373 et GC376 pour le SARS-CoV-2 Mpro et c Clivage SARS-CoV Mpro de Abz-SVTLQSG-Y(NO2)-R. N = 3, les valeurs sont représentées par la moyenne ± SE.
I
Tableau 1 Paramètres catalytiques du SARS-CoV Mpro et du SARS-CoV-2 Mpro.
Tableau complet
Table 1 Catalytic parameters of SARS-CoV Mpro and SARS-CoV-2 Mpro.
From: Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication
| Protease | K0.5 (µM) | kcat (min−1) | kcat /K0.5 (min−1 µM−1) | Hill coefficient |
|---|---|---|---|---|
| SARS-CoV-2 Mpro | 70 ± 10 | 135 ± 6 | 1.8 ± 0.4 | 2.2 |
| SARS-CoV Mpro | 52 ± 17 | 30 ± 2 | 0.6 ± 0.2 | 1.9 |
Les mesures de la CI50 ont révélé que la GC373 et la GC376 inhibent toutes deux le SARS-CoV Mpro et le SARS-CoV-2 Mpro in vitro à des concentrations nanomolaires (Fig. 1b, c). Pour le CoV-Mpro-SARS-2, les CI50 pour la GC373 et la GC376 sont respectivement de 0,40 ± 0,05 μM et de 0,19 ± 0,04 μM. Ceci est en accord avec les études du GC376 avec le Mpro de virus apparentés. Pour le FCoV Mpro, la CI50 pour le GC376 était de 0,49 ± 0,07 μM, tandis que la CI50 pour le GC376 pour le Mpro des coronavirus du furet et du vison était de 1,33 ± 0,19 μM et de 1,44 ± 0,38 μM, respectivement12. Pour le Mpro du coronavirus du SRAS, nous avons observé une CI50 accrue, démontrant une large inhibition par les deux composés, la GC373 et la GC376 étant respectivement de 0,070 ± 0,02 μM et de 0,05 ± 0,01 μM. Le produit d’addition du bisulfite GC376 présente une puissance légèrement supérieure pour les deux enzymes par rapport à l’aldéhyde libre. Nos valeurs in vitro de la CI50 pour le GC373 et le GC376 reflètent une liaison étroite pour le CoV-2 Mpro du SRAS par rapport à d’autres inhibiteurs testés in vitro, par exemple, ebselen (CI50 0,67 μM)16, tideglusib (CI50 1,55 μM)16, carmofur (CI50 1,82 μM)16, disulfirame (CI50 9,35 μM)16, shikonine (CI50 15,75 μM)16, PX-12 (CI50 21,39 μM)16. Le GC373 et le GC376 sont également plus puissants que les inhibiteurs de cétoamide récemment signalés (la CI50 du composé principal est de 0,67 μM)15. Récemment, un inhibiteur peptidylique apparenté a été signalé avec une ogive similaire à celle de notre composé, mais avec un groupe indole en position P3 et une CI50 de 0,05 ± 0,005 μM18. Cependant, ce composé n’a pas été démontré comme étant efficace chez les animaux, comme c’est le cas pour le GC376.
Structure cristalline du SARS-CoV-2 Mpro en complexe avec la GC373 et la GC376
Pour mieux comprendre le mécanisme d’inhibition, les structures cristallines du Mpro du CoV-2-SARS avec les inhibiteurs GC373 et GC376 ont été déterminées à 2,0 et 1,9 Å, respectivement (Fig. 2 et Tableau supplémentaire 1), et comme prévu, le promédicament s’est converti en médicament, ce qui a donné des ligands identiques dans les structures. La structure tridimensionnelle de l’apo-SARS-CoV-2 Mpro (code PDB 6WTM) est très similaire aux structures récemment résolues avec un écart quadratique moyen de 0,19 Å (code PDB 6Y2E)15,16. Le SARS-CoV-2 Mpro a cristallisé sous la forme d’un dimère facilité par un doigt N du protomère A (résidus 1-7) qui s’insère dans une poche du protomère B. Chaque protomère présentait une structure à deux lobes dont l’un était composé de deux barillets β antiparallèles (domaines I et II), qui forment un pli de chymotrypsine et de peptidase de type 3C, le site actif étant constitué d’une dyade Cys145-His41 située à l’interface du domaine. Le trou oxyanionique, influencé par la dimérisation19, est formé à partir des principaux résidus de la chaîne Gly143, Ser144 et Cys145. Le domaine C-terminal III est impliqué dans l’échange de domaines et facilite la formation du dimère20. Le remplacement moléculaire par la structure 6Y7M.PDB a révélé la densité électronique dans la carte Fo-Fc au niveau de la cystéine catalytique pour le promédicament GC376 (code PDB 6WTJ) et le médicament GC373 (code PDB 6WTK). Dans les deux structures, l’inhibiteur peptidylique est fixé de manière covalente à Cys145 sous forme d’hémiacétal, ce qui montre que le groupe bisulfite quitte la GC376 (Fig. supplémentaire 17
Contrairement à la structure MERS Mpro-GC376, la densité d’électrons Mpro du SARS-CoV-2 a indiqué la formation d’un seul énantiomère pour cet inhibiteur14. Les contributions de Gly143, Ser144 et Cys145 au squelette définissent le trou d’oxyanion. Le trou d’oxyanion est stabilisé par un tour de β où le carbonyle de l’hydrogène du Leu141 se lie à l’amine du squelette du Ser144, et l’amide du squelette du Leu141 interagit avec l’hydroxyle du Ser144 (Fig. 3a). Les interactions hydrophobes et un réseau de liaisons hydrogène sont tous deux établis par les résidus du domaine II pour favoriser la liaison de l’inhibiteur. P2 s’insère dans une poche hydrophobe composée de la base générale His41 et des résidus Met49 et Met165, représentant le sous-site S2 de l’enzyme. (Fig. 3b). Le substitut de glutamine en position P1 du substrat forme une liaison hydrogène avec la chaîne latérale de His163 et Glu166 et une interaction hydrophobe avec His172. Le carbonyle en position P3 forme une liaison hydrogène avec l’amide de l’épine dorsale de Glu166 (Fig. 3c, d). Comme ce qui a été observé dans la structure MERS Mpro-GC37614, le cycle benzyle et le γ-lactame du substitut Gln forment une interaction hydrophobe empilée, qui stabilise l’inhibiteur dans le site actif de la protéase. Ensemble, cela permet une forte liaison et une faible CI50 pour l’inhibiteur. Un examen attentif du sous-site de la Mpro du CoV-2-SARS révèle des régions permettant le développement futur de l’inhibiteur (Fig. supplémentaire 17).
Fig. 2 : La structure cristalline du SARS-CoV-2 Mpro en complexe avec le GC373 (converti à partir du GC376).
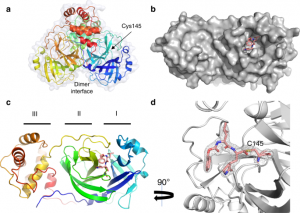
a L’Apo-SARS-CoV-2 Mpro forme un dimère (6WTM.pdb). b Le promédicament GC376, lorsqu’il est incubé avec le SARS-CoV-2 Mpro, se transforme en GC373 qui forme une liaison covalente avec C145. La représentation de la surface révèle la poche du site actif dans le complexe avec le GC373 (6WTJ.pdb). c Représentation en ruban d’un protomère du SARS-CoV-2 dans le complexe avec l’inhibiteur GC373 se liant au domaine II. d Le GC373 interagit de manière covalente avec la cystéine du site actif du SARS-CoV-2 Mpro. La densité électronique sur 1σ est indiquée en grisé.
Fig. 3 : Le GC373 se fixe dans la poche de site active du SARS-CoV-2 Mpro.
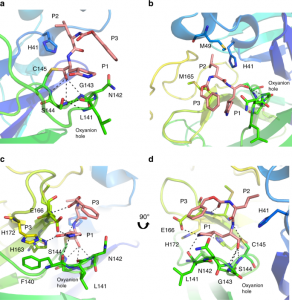
a La GC373 forme une liaison covalente avec C145, et l’oxyanion est stabilisé par des liaisons H du squelette avec G143, S144, et C145. b La position P2 de la GC373 est stabilisée par une poche hydrophobe par la base générale H41, et les résidus M49, et M165. c, d Des liaisons H sont établies avec la GC373 et les chaînes latérales de H163 et E166, ainsi que le squelette du résidu E166. Le SARS-CoV-2 Mpro est représenté en dessin animé avec l’inhibiteur en rose. P1, P2 et P3 de l’inhibiteur peptidylique sont indiqués. Code PDB : 6WTJ.
Pour confirmer la formation d’un hémi-acétal covalent comme énantiomère unique dans le site actif, le GC373 a été préparé avec un marquage 13C (>99%) sur le carbone aldéhydique et mélangé en excès de 7,8 fois avec la protéase Mpro du SARS-CoV-2 dans un tampon deutéré. L’analyse RMN du HSQC (700 MHz) a montré l’apparition d’un seul signal de crête croisée (un seul isomère) pour le carbone de l’hémiacétal à 76 ppm (13C) et 5,65 ppm (1H) conformément aux précédents rapports de déplacement chimique pour les hémiacétals7 (Fig. 18 supplémentaire).
Le SARS-CoV-2 Mpro a une activité catalytique accrue par rapport au SARS-CoV Mpro
Une analyse récente de la structure cristalline a révélé des différences dans les résidus résidant entre l’interface dimère du SARS-CoV-2 Mpro et celle du SARS-CoV Mpro 15. Des études de mutagénèse antérieures, qui ont modifié les résidus à l’interface dimère du SARS-CoV Mpro, et plus particulièrement une variante T285A, ont augmenté l’activité catalytique de 3,6 fois21. Dans le SARS-CoV-2, le T285 est une alanine, et en accord avec cela, notre analyse montre que le taux de renouvellement catalytique du SARS-CoV-2 Mpro (135 ± 6 min-1) est presque cinq fois plus rapide que celui du SARS-CoV Mpro (30 ± 2 min-1) avec notre substrat Abz-SVTLQSG-TyrNO2R (tableau 1). Avec ce substrat FRET, nous démontrons une efficacité catalytique supérieure avec le SARS-CoV-2 Mpro (1,8 ± 0,4 min-1 µM-1) par rapport au SARS-CoV Mpro (0,6 ± 0,2 min-1 µM-1). Cette conclusion contraste avec les rapports récents où aucune différence n’a été observée dans l’efficacité catalytique entre le SARS-CoV Mpro et le SARS-CoV-2 Mpro en utilisant un substrat différent : 3011 ± 294 s-1 M-1 (0,18 ± 0,02 min-1 µM-1) et 3426 ± 416,9 s-1 M-1 (0,2 ± 0,03 min-1 µM-1) respectivement15. Il reste à déterminer si cela influence la virulence du CoV-2 du SRAS.
Les GC373 et GC376 sont de puissants inhibiteurs du CoV-2 du SRAS en culture cellulaire
Pour tester la capacité des GC373 et GC376 à inhiber le CoV-2 du SRAS, des tests de réduction de la plaque ont été effectués sur des cellules Vero E6 infectées en l’absence ou en présence de concentrations croissantes de GC373 (Fig. 4a) ou de GC376 (Fig. 4b) pendant 48 h. Les résultats ont été présentés sous forme de pourcentage d’inhibition du nombre d’unités formatrices de plaque par puits. La CE50 pour le GC373 était de 1,5 μM, alors que la CE50 pour le GC376 était de 0,92 μM. Pour examiner la cytotoxicité des cellules, la production d’ATP a été mesurée à l’aide du test CellTiter-Glo sur des cellules Vero E6 ou A549 incubées en présence des inhibiteurs pendant 24 h. Les valeurs de CC50 pour la GC373 et la GC376 étaient >200 μM. Afin d’examiner plus en détail l’activité antivirale des GC373 et GC376 dans un essai à large gamme dynamique, un essai de réduction du rendement du virus a été réalisé. Une RT-PCR (reverse transcription polymerase chain reaction) quantitative a été réalisée sur des surnageants de cellules non traitées, et des cultures de cellules traitées par GC373- (Fig. 4c) ou GC376- (Fig. 4d) infectées à une multiplicité d’infection (MOI) = 0,01 pendant 48 h. Il a été observé que GC373 et GC376 sont tous deux de puissants inhibiteurs de SARS-CoV-2, diminuant les titres viraux de valeurs de 3 log, par rapport à une diminution de valeur de 2 log dans les résultats récemment publiés utilisant d’autres composés aldéhydiques18. Ces résultats indiquent que le GC373 et le GC376 sont tous deux de puissants inhibiteurs du CoV-2 du SRAS, avec un indice thérapeutique supérieur à 200.
Fig. 4 : Les GC373 et GC376 inhibent puissamment le CoV-2 du SRAS.
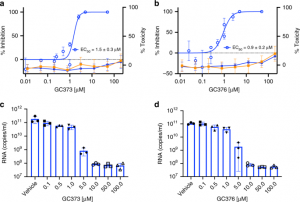
a, b La croissance du CoV-2 du SRAS dans les cellules Vero E6 a été déterminée par des tests en plaque 48 heures après l’infection en présence de diverses concentrations de médicament. La cytotoxicité a été mesurée à l’aide du test CellTiter-Glo. a Pourcentage d’inhibition du SARS-CoV-2 par GC373 dans les cellules Vero E6 (cercles bleus ouverts) et cytotoxicité dans les cellules Vero E6 (cercles bleus fermés) et A549 (orange). b Pourcentage d’inhibition du SARS-CoV-2 par GC376 dans les cellules Vero E6 (cercles bleus ouverts) et cytotoxicité dans les cellules Vero E6 (bleus, cercles fermés) et A549 (orange). c, d Pour mesurer la réduction du rendement du virus, les cellules Vero E6 ont été infectées avec MOI = 0,01 de SARS-CoV-2 en triple exemplaire sans ou avec différentes concentrations de GC373 c ou GC376 d pendant 24 h et les surnageants ont été récoltés, l’ARN isolé et quantifié par qRT-PCR. Dans les courbes EC50, les valeurs sont représentées par la moyenne ± SD d’au moins deux expériences indépendantes. Pour le test de toxicité, les valeurs sont représentées par la moyenne ± l’écart-type de 12 expériences indépendantes. Les données sur l’ARN sont présentées sous forme de moyenne ± ÉT, avec n = 3 expériences indépendantes et des points de données individuels indiqués.
Discussion
De nombreux médicaments ont été conçus à l’origine pour inhiber le CoV Mpro3 du SRAS, mais l’épidémie de SRAS de 2002 a été contrôlée par des mesures de santé publique et ces composés n’ont jamais été homologués. Le GC376 a été utilisé pour traiter avec succès le FIP chez les chats car son produit de dégradation, le GC373, inhibe efficacement le Mpro du FCoV. Des analogues de ces médicaments ont également inhibé le Mpro du CoV MERS et bloqué la réplication virale dans les cellules à une EC50 de 0,5 μM14. Nos études montrent que le GC376 et le GC373 sont des inhibiteurs efficaces du Mpro du CoV-2 du SRAS, en accord avec des études antérieures montrant que le GC373 et le GC376 possèdent une large spécificité contre le Mpro des coronavirus et des calcivirus (Tableau supplémentaire 2)10,13,14,22. Il est clair que ces médicaments doivent être rapidement avancés dans les essais sur l’homme pour COVID-19. Le SRAS-CoV-2 est la cause de COVID-19 et est un virus avec un taux de mutation important23. En outre, chez certains patients, le virus a persisté plus de deux mois avec une possibilité de réinfection24. Les vaccins sont d’une importance capitale, mais il est probable qu’ils ne seront pas disponibles avant un an ou plus, car ce virus peut poser des problèmes de vaccination.
Il existe de nombreux essais cliniques qui testent des médicaments dont les indications initiales ont été modifiées. Le remdesivir, un inhibiteur de la polymérase mis au point pour traiter le virus Ebola25, avait initialement été utilisé dans des cas de compassion, et est maintenant approuvé par la FDA pour une utilisation d’urgence chez les patients atteints de COVID-1926. L’EIDD 2801 (un triphosphate de N4-hydroxycytidine qui est incorporé dans l’ARN viral pour favoriser les erreurs dans l’ARN de la progéniture) est un autre médicament conçu pour inhiber l’ARN polymérase dépendante de l’ARN, y compris dans les coronavirus. Ces exemples d’antiviraux à action directe (DAA) pour COVID-19 sont d’une importance capitale27. Le GC373 et le GC376 sont également des AAD conçus spécifiquement pour les coronavirus. Il est probable que plusieurs médicaments très puissants seront nécessaires en combinaison pour traiter le SRAS-CoV-2 et pour prévenir l’amplification des mutants résistants aux médicaments28, comme ce fut le cas pour le VIH29. Les résultats de cette étude montrent que le GC373 et le GC376 sont des antiviraux candidats qui devraient être accélérés dans les essais cliniques pour le COVID-19.
Méthodes
Synthèse de GC373, GC376, et du substrat peptidique FRET
Les protocoles, les détails synthétiques, la caractérisation des composés et les informations méthodologiques connexes sont présentés dans les méthodes complémentaires. L’identité et la pureté (>95%) de tous les composés ont été déterminées par chromatographie (silice ou HPLC RP-18), avec attribution complète des spectres RMN 1H et 13C, et des spectres de masse à haute résolution.
Clonage, expression et purification de SARS-CoV Mpro et SARS-CoV-2 Mpro
L’ADN codant pour la principale protéase Mpro du SARS-CoV-2 (Genbank : MN908947.3) a été obtenu auprès de BioBasic Inc. (Ontario, Canada) et contient un site de restriction EcoRI en amont et un site de restriction HindIII en aval du gène de fusion. Le gène Mpro a été optimisé par un codon pour l’expression dans Escherichia coli. Le gène de fusion a été cloné dans les sites de restriction EcoRI et HindIII du vecteur d’expression (Invitrogen) pET SUMO (small ubiquitin-like modifier), ce qui garantit que la protéine Mpro du SRAS-CoV-2 est en phase avec la protéine SUMO marquée His. Le plasmide résultant a été transformé en E. coli BL21(DE3), induit avec 0,5 mM d’isopropyle β-d-1-thiogalactopyranoside et la protéine a été exprimée pendant 4-5 h à 37 °C. Les cellules ont été récoltées par centrifugation (4400 × g pendant 10 min à 4 °C) et mises en suspension dans un tampon de lyse (20 mM Tris-HCl pH 7,8, 150 mM NaCl, 5 mM imidazole). Les cellules ont été lysées par sonication, les débris cellulaires ont été éliminés par centrifugation (27 000 × g pendant 10 min à 4 °C), et le surnageant a été chargé sur une colonne de résine Ni-NTA (Qiagen). Après avoir lavé la résine avec 10 volumes de colonne de tampon de lyse contenant 20 mM d’imidazole, la protéine de fusion a été éluée avec 40-500 mM d’imidazole dans le tampon de lyse. Les fractions éluées ont été analysées par électrophorèse sur gel de dodécylsulfate de sodium-polyacrylamide (SDS-PAGE), les échantillons avec la protéine d’intérêt ont été regroupés, dialysés contre un tampon de lyse contenant 1 mM de TNT à 4 °C et concentrés en utilisant un filtre Amicon Ultra-15 (Millipore) avec un MWCO de 10 kDa. La protéine de fusion a été digérée avec la protéase SUMO marquée par His (McLab, South San Francisco, CA) à 4 °C pendant 1 à 2 heures pour éliminer la marque SUMO. Le mélange de clivage a ensuite été passé à travers une colonne de résine Ni-NTA. Le flux contenant le SARS-CoV-2 Mpro a été collecté et analysé par SDS-PAGE. La protéine SARS-CoV-2 Mpro a ensuite été purifiée en utilisant une colonne de chromatographie d’exclusion de taille (G-100, GE Healthcare,) équilibrée avec 20 mM Tris, 20 mM NaCl, 1 mM DTT, pH 7,8. Les fractions contenant la protéine Mpro SARS-CoV-2 ont été mises en commun et concentrées à l’aide d’un filtre Amicon Ultra-15 avec un MWCO de 10 kDa. En outre, un échantillon de la protéine de fusion SUMO-SARS-CoV-2 Mpro a également été purifié par chromatographie d’exclusion de taille et concentré comme décrit ci-dessus. Le plasmide codant pour la protéine Mpro du SRAS-CoV avec un His-tag N-terminal en amont d’un site de clivage du FactorX est un cadeau du Dr Michael James. La protéine a été exprimée et purifiée selon le protocole précédent9.
Spectrométrie de masse du SARS-CoV-2 Mpro
La masse du SARS-CoV-2 Mpro libre a été confirmée par HR-MALDI sur un MALDI-TOF (Bruker Ultrafelxtreme, Bruker Daltronics, USA) et LC-MS sur un instrument ESI-TOF (Agilent Technologies 6220, CA, USA) en utilisant l’ionisation par électrospray et analysée avec le logiciel d’analyse qualitative Agilent Mass Hunter (version B.03.01 SP3).
Cinétique enzymatique du SARS-CoV-2 et du SARS-CoV Mpro
Un substrat fluorescent synthétisé contenant le site de clivage (indiqué par la flèche, ↓) du SARS-CoV-2 Mpro (Abz-SVTLQ↓SG-Tyr(NO2)-R) a été utilisé pour le test de clivage basé sur le transfert d’énergie de résonance de fluorescence (FRET)15. Les réactions de protéase du SARS-CoV-2 Mpro et du SARS-CoV Mpro vers le substrat fluorescent ont été effectuées dans un tampon d’activité (20 mM Bis-Tris, pH 7,8, 1 mM DTT) à 37 °C pendant 10 minutes. La concentration finale des protéases utilisées dans l’essai a été fixée à 80 nM et les concentrations du substrat ont varié de 0,1 à 500 μM. La réaction a commencé avec l’enzyme et le signal de fluorescence du produit de clivage peptidique Abz-SVTLQ a été contrôlé à une longueur d’onde d’émission de 420 nm avec excitation à 320 nm, en utilisant un spectrophotomètre de fluorescence Flx800 (BioTek). Avant les calculs cinétiques, il a été vérifié que la proportionnalité entre la fluorescence émise et la quantité de substrat utilisée dans l’essai était linéaire. La concentration minimale de l’enzyme et le temps de réaction qui donnaient une dépendance linéaire de la quantité de produit généré avec le temps ont été choisis. Les vitesses initiales en unités de fluorescence relatives correspondantes par unité de temps (ΔRFU/s) ont été converties en quantité de substrat clivé par unité de temps (μM/s) par ajustement à la courbe d’étalonnage de l’Abz-SVTLQ libre. Toutes les données ont été corrigées des effets du filtre interne par un protocole adopté dans la littérature17. En bref, le signal de fluorescence (RFU) à chaque concentration de substrat a été déterminé et défini comme f(FRET). Ensuite, 5 μL d’Aminobenzoyl-SVTLQ libre à la concentration finale de 5 μM ont été ajoutés à chaque concentration de substrat et la fluorescence a été mesurée : f(FRET + Aminobenzoyl-SVTLQ). Simultanément, une lecture de référence a été prise avec la même concentration d’Aminobenzoyl-SVTLQ libre et a été définie comme f(ref). La correction du filtre interne a été obtenue comme suit : corr% = (f (FRET + aminobenzoyl-SVTLQ) – f (FRET))/f (ref) × 100%. La vitesse initiale corrigée de la réaction a été calculée comme suit : v = vo/(corr%). vo représente la vitesse initiale de chaque réaction.
Les paramètres cinétiques (Vmax et Km) ont été dérivés en ajustant le tracé des vitesses initiales corrigées en fonction des concentrations de substrat à l’équation de Michaelis-Menten, v = vmax × [S]/(Km + [S]) en utilisant le logiciel GraphPad Prism (GraphPad 8.3.1). kcat/Km a été calculé selon l’équation, kcat/Km = vmax/([E] × Km). Des expériences triples ont été effectuées pour chaque point de données, et la valeur a été présentée comme moyenne ± écart-type (SD).
Paramètres d’inhibition
Les solutions mères de GC373 et GC376 ont été préparées avec du diméthyl sulfoxyde (DMSO) aqueux à 10 %. Pour la détermination de la CI50, 80 mM de SARS-CoV-2 Mpro ont été incubés avec la GC373 ou la GC376 à différentes concentrations de 0 à 100 μM dans 20 mM de Bis-Tris, pH 7,8, 1 mM de DTT à 37 °C pendant 10 min. La réaction protéasique a été déclenchée par l’ajout de 100 μM du substrat. Le logiciel GraphPad Prism (GraphPad 8.3.1) a été utilisé pour le calcul des valeurs de la CI50. Les deux inhibiteurs ont été testés pour une liaison non spécifique en effectuant un titrage de référence en l’absence de TNT ne montrant aucune influence dans les lectures de fluorescence obtenues (données non montrées).
Cristallisation
Pour la cristallisation, le SARS-CoV-2 Mpro purifié a été dialysé contre un tampon contenant 10 mM de NaCl et 5 mM de Tris-HCl pH 8,0 pendant la nuit à 4 °C, et concentré avec un filtre centrifuge Millipore (seuil de coupure de 10 kDa MW) à 9 mg/mL. La protéine a été incubée avec un excès de cinq molaires d’inhibiteur à 4 °C pendant 2 h avant la cristallisation. Pour le SARS-CoV-2 Mpro, la protéine a été soumise à l’écran de cristallisation PACT (Molecular Dimensions), avec des succès identifiés dans plusieurs conditions pour les deux inhibiteurs. Pour l’Apo- SARS-CoV-2 Mpro, les meilleurs cristaux ont été observés avec des plateaux de gouttes suspendus à température ambiante dans un rapport de 1:1 avec de la liqueur mère 0,2 M de sulfate de sodium, 0,1 M de Bis-Tris propane pH 6,5, et 20% p/v de PEG 3350. Le SARS-CoV-2 Mpro avec inhibiteurs a cristallisé avec une liqueur mère contenant du chlorure de sodium 0,2 M, de l’acide 4-(2-hydroxyéthyl)-1-pipérazine-éthanesulfonique (HEPES) pH 7,0, et 20 % p/v de PEG 6000. Avant la congélation, les cristaux ont été incubés avec 15 % de glycérol comme cryoprotecteur. Les cristaux ont d’abord été criblés sur notre source domestique 007 MicroMax (Rigaku Inc) avec criblage des cristaux et collecte finale des données à la source de rayonnement synchrotron de Stanford (SSRL), ligne de faisceau 12-2 avec Blu-Ice en utilisant l’interface Web-Ice30.
Collecte de données sur la diffraction, détermination de la phase, construction de modèles et affinement
Tous les ensembles de données de diffraction ont été recueillis à l’aide d’un rayonnement synchrotron de longueur d’onde 0,97946 Å à la ligne de faisceaux 12-2 du SSRL California, aux États-Unis, en utilisant un détecteur Dectris PILATUS 6 M. Plusieurs ensembles de données ont été recueillis à partir des cristaux de l’enzyme libre SARS-CoV-2 Mpro ainsi qu’avec les GC376 et GC373 traitées. Le XDS231 et le Scala ont été utilisés pour le traitement des ensembles de données. L’ensemble de données de diffraction de l’enzyme libre SARS-CoV-2 Mpro a été traité à une résolution de 1,75 Å, dans le groupe spatial P21 (tableau supplémentaire 1). Pour le complexe de SARS-CoV-2 Mpro avec GC376 et GC373, l’ensemble de données collectées a été traité avec une résolution de 1,9 Å et 2,0 Å et dans le groupe spatial C2 (Tableau supplémentaire 1). Les trois structures ont été déterminées par remplacement moléculaire avec la structure cristalline de l’enzyme libre du SARS-CoV-2 Mpro (entrée PDB 6Y7M) comme modèle de recherche, en utilisant le programme Phaser de Phenix32, version v1.18.1-3855). Le Ligand Fit de Phenix a été utilisé pour l’ajustement des deux inhibiteurs dans la densité de la carte pré-calculée à partir du raffinement de Phenix, en utilisant le code de ligand K36. L’affinement des trois structures a été réalisé avec phenix.refine dans le logiciel Phenix. Les statistiques de diffraction, de traitement des données et de raffinement du modèle sont données dans (tableau supplémentaire 1). Le modèle a été inspecté avec des tracés de Ramachandran et les modèles finaux ont été affichés à l’aide du logiciel de graphiques moléculaires PyMOL (version 1.7.6.5 Schrödinger, LLC).
Détermination de la CE50 par le test de réduction en plaque
SARS-CoV-2/CANADA/VIDO 01/2020 est un aimable cadeau de Darryl Falzarano (Université de Saskatchewan). Les cellules E6 de Vero (rein de singe vert femelle) ont été infectées avec un MOI de 0,0001 pfu/cellule dans un milieu d’infection composé du milieu de Dulbecco’s Modified Eagle’s complété par 1× acides aminés non essentiels (Gibco), 10 mM HEPES, 2% de sérum foetal bovin, 50 IU/mL de pénicilline, 50 IU/mL de streptomycine ; différentes doses de GC373 ou GC376. Après 1 h, le milieu infectant a été retiré et les monocouches ont été recouvertes d’un milieu de croissance (MEM complété par 10 mM HEPES) contenant 1,2% d’Avicel RC-591 (DuPont), contenant la dose correspondante de GC373 ou GC376. Après 48 h, les cellules ont été fixées dans 10 % de formaldéhyde, et colorées avec 0,5 % (p/v) de cristal violet. Les plaques ont été comptées et les données ont été tracées en % d’inhibition par rapport au log10 [médicament] en utilisant le Prism (GraphPad). Les valeurs de la CE50 ont été déterminées à l’aide d’une analyse de régression non linéaire. Les expériences ont été réalisées en trois exemplaires. Les barres d’erreur indiquent l’écart-type. Pour examiner la réduction de la sécrétion d’ARN viral par les cellules Vero E6 causée par la GC373 et la GC376, les cellules ont été infectées par le SRAS-CoV-2 MOI = 0,01 pfu/cellule en présence de concentrations variables de GC373 ou GC376, pendant 1 h, le milieu a été retiré et remplacé par un milieu de croissance contenant également la GC373 et la GC376 et des surnageants récoltés 48 h plus tard et quantifiés par RT-PCR quantitative.
Quantification de l’ARN viral du SRAS-CoV-2 dans les surnageants de culture cellulaire par RT-PCR quantitative
Des surnageants cellulaires (140 µL) ont été collectés à différents points après l’infection, et l’ARN a été isolé à l’aide du mini kit d’ARN viral QIAmp selon les instructions du fabricant (Qiagen). La transcription inverse a été effectuée sur 2 µL en utilisant le mélange maître Superscript IV Vilo (Invitrogen). La PCR quantitative a été réalisée en utilisant 2 µl d’ADNc dans le mélange maître TaqMan Fast en utilisant des amorces et une sonde pour le gène N (amorces N2) conçues par le centre américain de contrôle et de prévention des maladies (IDT cat#10006606). Une courbe standard a été générée en utilisant des dilutions de standards de contrôle positif du CDC (IDT cat # 10006625).
Mesure de la cytotoxicité dans les cellules A549 et Vero E6
La viabilité cellulaire a été mesurée à l’aide du test de viabilité cellulaire luminescent CellTiter-Glo (Promega). Des cellules A549 (épithélium pulmonaire humain mâle) ou des cellules Vero E6 ont été ensemencées à 5 × 103 cellules/puits dans des plaques de 96 puits et incubées pendant la nuit avant le traitement. Les composés GC373 et GC376 ont été solubilisés dans du DMSO et ajoutés aux cellules dans une dilution en série de huit points et quatre fois (200 µM à 0,0122 µM). Les cellules ont été incubées en présence des composés pendant 24 heures avant l’ajout du substrat luminescent et la mesure de l’activité de l’ATP selon les instructions du fabricant. Le pourcentage de cellules viables a été calculé par rapport aux cellules traitées avec le solvant seul (0,5 % de DMSO). Les résultats ont été tracés comme la moyenne de trois expériences indépendantes ±SD, où chaque expérience consistait en quatre puits par concentration de composé.
Résumé du rapport
De plus amples informations sur la conception de la recherche sont disponibles dans le résumé du rapport de recherche sur la nature lié à cet article.
Disponibilité des données
____________________________________________________________________________________________Les auteurs déclarent que les données à l’appui des conclusions de cette étude sont disponibles dans les fichiers de l’article, des données supplémentaires et de la méthode supplémentaire. Le modèle et la carte de la protéase du SRAS-CoV-2, sous forme apo, avec le médicament GC373, et avec le promédicament GC376, sont disponibles dans la banque de données sur les protéines du RCSB PDB 6WTM, 6WTK et 6WTJ respectivement. Les données RMN peuvent être consultées ici [http://deposit.bmrb.wisc.edu/author_view/BMRB/50262_hy_lssvgcxu.str]. Les données sources sont fournies avec le présent document.
Références [].
1. OMS. www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports (2020).
2. OMS. http://www.who.int/csr/sars/country/2003_07_11/en/ (2003).
3.Pillaiyar, T., Manickam, M., Namasivayam, V., Hayashi, Y. & Jung, S. H. An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3cl protease inhibitors : peptidomimetics and small molecule chemotherapy. J. Med Chem. 59, 6595–6628 (2016).
Article de la CAS Google Scholar
4.Otto, H. H. & Schirmeister, T. Cystéine protéases et leurs inhibiteurs. Chimie. Rev. 97, 133-172 (1997).
Article du CAS Google Scholar
5.Drag, M. & Salvesen, G. S. Emerging principles in protease-based drug discovery. Nat. Rev. Drug Discov. 9, 690–701 (2010).
Article de la CAS Google Scholar
6.Flexner, C., Bate, G. & Kirkpatrick, P. Tipranavir. Nat. Rev. Drug Discov. 4, 955–956 (2005).
Article de la CAS Google Scholar
7.Malcolm, B. A. et al. Peptide aldéhyde inhibiteurs de la protéinase 3C du virus de l’hépatite A. Biochemistry 34, 8172-8179 (1995).
Article CAS Google Scholar
8.Chang, K. O., Kim, Y., Lovell, S., Rathnayake, A. D. & Groutas, W. C. Antiviral drug discovery : norovirus proteases and development of inhibitors. Virus 11, 197 (2019).
9.Yin, J. et al. Une vue mécaniste de l’inhibition des enzymes et de l’hydrolyse des peptides dans le site actif de la peptidase semblable au SARS-CoV 3C. J. Mol. Biol. 371, 1060-1074 (2007).
Article du CAS Google Scholar
10.Kim, Y. et al. Inhibiteurs à large spectre contre les protéases de type 3C des coronavirus félins et des calicivirus félins. J. Virol. 89, 4942–4950 (2015).
Article du CAS Google Scholar
11.Pedersen, N. C. et al. Efficacité d’un inhibiteur de la protéase de type 3C dans le traitement de diverses formes de péritonite infectieuse féline acquise. J. Feline Med. Surg. 20, 378-392 (2018).
Article Google Scholar
12, K. D. et al. Inhibiteurs de protéase largement efficaces contre les coronavirus des félins, des furets et des visons. Antivir. Res. 160, 79-86 (2018).
Article du CAS Google Scholar
13.Kim, Y. et al. Antiviraux à large spectre contre les protéases 3C ou 3C-like des picornavirus, des norovirus et des coronavirus. J. Virol. 86, 11754–11762 (2012).
Article du CAS Google Scholar
14.Galasiti Kankanamalage, A. C. et al. Conception structurée d’inhibiteurs puissants et perméables de la protéase 3CL du coronavirus MERS qui utilisent une fraction pipéridine comme nouvel élément de conception. Eur. J. Med. Chem. 150, 334–346 (2018).
Article du CAS Google Scholar
15.Zhang, L. et al. La structure cristalline de la protéase principale du SRAS-CoV-2 fournit une base pour la conception d’inhibiteurs d’alpha-cétoamide améliorés. Science 368, 409-412 (2020).
16.Jin, Z. et al. Structure de la M(pro) du virus COVID-19 et découverte de ses inhibiteurs. Nature 582, 289-293 (2020).
17.Blanchard, J. E. et al. Le criblage à haut débit permet d’identifier les inhibiteurs de la protéinase principale du coronavirus du SRAS. Chimie. Biol. 11, 1445-1453 (2004).
Article du CAS Google Scholar
18.Dai, W. et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 368, 1331-1335 (2020).
19.Shi, J., Sivaraman, J. & Song, J. Mechanism for controlling the dimer-monomer switch and coupling dimerization to catalysis of the severe acute respiratory syndrome coronavirus 3C-like protease. J. Virol. 82, 4620–4629 (2008).
Article du CAS Google Scholar
20.Zhong, N. et al. Le domaine C-terminal de la protéase principale du SARS-CoV peut former un dimère à domaine échangé en 3D. Protein Sci. 18, 839-844 (2009).
CAS PubMed PubMed Central Google Scholar
21.Hu, T. et al. Deux mutations adjacentes sur l’interface dimère de la protéase de type 3C du coronavirus du SRAS provoquent des changements conformationnels différents dans la structure cristalline. Virology 388, 324-334 (2009).
Article du CAS Google Scholar
22.Kim, Y. et al. Inversion de la progression de l’infection mortelle par un coronavirus chez le chat grâce à un inhibiteur de la protéase du coronavirus à large spectre. Pathogène PLoS. 12, e1005531 (2016).
Article Google Scholar
23.Wang, C. et al. L’établissement d’une séquence de référence pour l’analyse du CoV-2 du SRAS et de ses variations. J. Med. Virol. 92, 667–674 (2020).
24.Biswas, A., et al. Emergence of Novel Coronavirus and COVID-19 : whether to stay or die out ? Crit. Rev. Microbiol. 46, 182–193 (2020).
25.Gordon, C. J. et al. Le remdesivir est un antiviral à action directe qui inhibe l’ARN polymérase ARN-dépendante du coronavirus 2 du syndrome respiratoire aigu sévère avec une puissance élevée. J. Biol. Chem. 295, 6785–6797 (2020).
26.Grein, J. et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 382, https://doi.org/10.1056/NEJMoa2007016 (2020).
27.Sheahan, T. P. et al. Un antiviral à large spectre biodisponible par voie orale inhibe le CoV-2 du SRAS dans des cultures de cellules épithéliales des voies respiratoires humaines et de multiples coronavirus chez la souris. Traduction scientifique. Méd. 12, eabb5883 (2020).
28.Deng, X. et al. Les coronavirus résistants à un inhibiteur de protéase de type 3C sont atténués pour la réplication et la pathogénèse, révélant une barrière génétique faible mais un coût d’adaptation élevé de la résistance. J. Virol. 88, 11886–11898 (2014).
Article Google Scholar
29.Menendez-Arias, L. Molecular basis of human immunodeficiency virus type 1 drug resistance : overview and recent developments. Antivir. Res. 98, 93-120 (2013).
Article de la CAS Google Scholar
30.Gonzalez, A. et al. Web-Ice : integrated data collection and analysis for macromolecular crystallography. J. Appl. Crystallogr. 41, 176-184 (2008).
Article du CAS Google Scholar
31.Kabsch, W. XDS. Acta Crystallogr. Sect. D. Biol. Crystallogr. 66, 125-132 (2010).
Article du CAS Google Scholar
32.Liebschner, D. et al. Macromomolecular structure determination using X-rays, neutrons and electrons : recent developments in Phenix. Acta Crystallogr. Sect. D. Struct. Biol. 75, 861-877 (2019).
Article CAS Google Scholar
Télécharger les références
Remerciements
Nous tenons à remercier le personnel de la source de lumière synchrotron de Stanford, en particulier le Dr Silvia Russi et Lisa Dunn. Nous remercions vivement le Dr Randy Whittal et Béla Reiz pour leur aide dans l’acquisition et l’interprétation des données de spectrométrie de masse. M.J.L, J.C.V. et D.L.T. remercient les IRSC et le CRSNG pour leur financement (COVID-19 SOF-549297-2019). D.L.T. remercie l’Institut de virologie Li Ka Shing et la chaire GSK de virologie pour leur soutien. W.V. a bénéficié d’une bourse d’études supérieures d’Alberta Innovates et T.L. d’une bourse du PCSG des IRSC. L’utilisation de la source de rayonnement synchrotron de Stanford, SLAC National Accelerator Laboratory, est soutenue par le Département américain de l’énergie, Office of Science, Office of Basic Energy Sciences sous contrat n°. DE-AC02-76SF00515. Le programme de biologie moléculaire structurelle du SSRL est soutenu par l’Office of Biological and Environmental Research du DOE, et par les National Institutes of Health, National Institute of General Medical Sciences (P41GM103393). Le contenu de cette publication relève de la seule responsabilité des auteurs et ne représente pas nécessairement les vues officielles des NIGMS ou des NIH.
Informations sur les auteurs
Affiliations
Département de chimie, Université de l’Alberta, Edmonton, T6G 2G2, AB, Canada
Wayne Vuong, Conrad Fischer, Tess Lamer, Ryan T. McKay, Marco J. van Belkum & John C. Vederas
Département de biochimie, Groupe de recherche sur les maladies protéiques des membranes, Université de l’Alberta, Edmonton, T6G 2R3, AB, Canada
Muhammad Bashir Khan, Elena Arutyunova, Howard S. Young & M. Joanne Lemieux
Département de microbiologie et d’immunologie médicales, Université de l’Alberta, Edmonton, T6G 2R3, AB, Canada
Justin Shields, Holly A. Saffran, Michael A. Joyce & D. Lorne Tyrrell
Institut de virologie Li Ka Shing, Université d’Alberta, Edmonton, T6G 2E1, AB, Canada
Justin Shields, Holly A. Saffran, Michael A. Joyce & D. Lorne Tyrrell
Contributions
.C.V., W.V., et T.L. ont contribué à la synthèse des inhibiteurs. W.V. a contribué à la synthèse du substrat FRET. M.J.v.B. a contribué au clonage. E.A., M.J.v.B., et C.F. ont contribué à la protéine purifiée. C.F. et E.A. ont contribué à la cinétique des enzymes. M.J.L., H.S.Y., E.A. et M.B.K. ont contribué à la cristallisation et à la détermination de la structure. W.V. et R.T.M. ont contribué aux études de RMN marquées. Le D.L.T., le M.A.J., le H.A.S. et le J.A.S. ont contribué aux études d’inhibition virale et de toxicité cellulaire. M.J.L. a rédigé le projet initial. Tous les auteurs ont lu et approuvé le manuscrit.
Auteurs correspondants
Correspondance avec D. Lorne Tyrrell ou John C. Vederas ou M. Joanne Lemieux.
Déclarations éthiques
Des intérêts concurrents
Les auteurs ne déclarent aucun intérêt concurrent.
Informations complémentaires
Informations sur l’évaluation par les pairs Nature Communications remercie Henry van den Bedem et les autres évaluateurs anonymes pour leur contribution à l’évaluation par les pairs de ce travail.
Note de l’éditeur Springer Nature reste neutre en ce qui concerne les revendications de compétence dans les cartes publiées et les affiliations institutionnelles.
Informations complémentaires
Droits et autorisations
Libre accès Cet article est publié sous une licence internationale Creative Commons Attribution 4.0, qui autorise l’utilisation, le partage, l’adaptation, la distribution et la reproduction sur tout support ou format, à condition que vous mentionniez correctement le nom de l’auteur ou des auteurs originaux et la source, que vous fournissiez un lien vers la licence Creative Commons et que vous indiquiez si des modifications ont été apportées. Les images ou autres matériels de tiers dans cet article sont inclus dans la licence Creative Commons de l’article, sauf indication contraire dans une ligne de crédit du matériel. Si le matériel n’est pas inclus dans la licence Creative Commons de l’article et que l’utilisation prévue n’est pas autorisée par la réglementation légale ou dépasse l’utilisation autorisée, vous devrez obtenir l’autorisation directement du détenteur du droit d’auteur. Pour consulter une copie de cette licence, visitez le site http://creativecommons.org/licenses/by/4.0/.
Réimpressions et autorisations
A propos de cet article
Vérifier la monnaie et l’authenticité via CrossMark
Citer cet article
Vuong, W., Khan, M.B., Fischer, C. et al. Un médicament à base de coronavirus félin inhibe la principale protéase du CoV-2 du SRAS et bloque la réplication du virus. Nat Commun 11, 4282 (2020). https://doi.org/10.1038/s41467-020-18096-2
Télécharger la citation
Reçu le 29 avril 2020
Accepté le 23 juillet 2020
Publié le 27 août 2020
DOIhttps://doi.org/10.1038/s41467-020-18096-2
Partager cet article
Toute personne avec qui vous partagez le lien suivant pourra lire ce contenu :
Fourni par l’initiative de partage de contenu Springer Nature SharedIt
Commentaires
En soumettant un commentaire, vous acceptez de respecter nos conditions et nos lignes directrices communautaires. Si vous trouvez quelque chose d’abusif ou qui ne respecte pas nos conditions ou lignes directrices, veuillez le signaler comme étant inapproprié.


